
Replication is the lifeblood of science – but how do you replicate when your cohort is the biggest available? EDiT Lab Associate Member and genomics crank-turner postdoc extraordinaire Joni gives three ways how.

There is little more important than replication in science. While we might lust over novelty and fetishise the unexpected, it’s the confirmation of the known that actually moves science forward. Traditionally, that has meant that you find something, you ask your frenemy with some similar data to see if they see the same thing, you confirm your finding, and then you get all the power and wealth and everyone wants to hire you. Or, more normally, they don’t see anything of the sort, you swear loudly, sulk for a day and then spend the next few weeks trying to understand exactly what’s gone wrong.

I have no idea who these scientists are, but they’re probably laughing at your failure to replicate that finding
But what do you do when there are no frenemies left? Not because you have finally completed your nefarious plan for world domination (Gerome), but because all the frenemies are now friends, and all the data are now one. In short, what do you do when there is no replication cohort? This, to an extent, is the situation that many of the investigators interested in psychiatric disorders now find themselves in. Many of the available genetic data on psychiatric disorders is now gathered together by the Psychiatric Genetics Consortium. So, how can you test whether your findings are real? Here’s three ways how…
1. Look within
In 2009, the International Schizophrenia Consortium had a slight problem. They had expended a great deal of time and effort to gather a large sample of individuals with schizophrenia and had consciously created a carefully-curated control cohort (which isn’t easy to say with a mouthful of beans). But, for all their work, they hadn’t quite got the punchline – they had found only a single genetic region (albeit one that is quite important) that differed between cases and controls at a level greater than chance (given that they had searched the whole genome), and the difference observed was only very slight.
So how to prove that this fledgling “genome-wide association study” (GWAS) thing for which they’d been pushing wasn’t a wash, and that, against all the slings and arrows of outrage fortune, the field should persevere? To do so, they developed the concept of “leave one out polygenic risk scoring”. Rather than examining how variants differ between cases and controls in all samples (call them ABCDEFG), they instead only looked at a sub-sample (ABCDEF) Then, they took the results from that sub-sample analysis and looked at whether weighting variants in the rest of the cohort (G) by the ABCDEF results gave a significant prediction. And it did – the genetic differences seen in one part of the cohort were (overall if not individually) mirrored in the other, independent part. Furthermore, this happened in all of the seven possible set-ups, suggesting that the overall effect was likely to be robust in entirely independent cohorts. And so, a crucial technique for internal replication was born, along with a whole field…
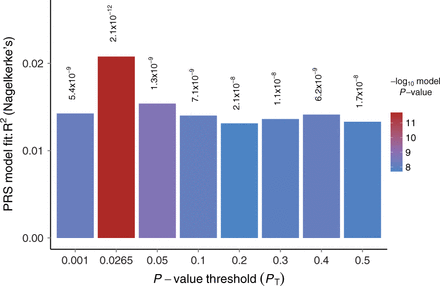
A Polygenic Risk Scoring Plot (Euesden et al, 2015: https://academic.oup.com/bioinformatics/article/31/9/1466/200539)
2. Squint a bit
At the beginning of the year, a big GWAS of IQ was published, identifying 18 parts of the genome where variants differ with increasing IQ. This was not the first such GWAS, but it represented a big step forward in the number of regions identified. But furthermore, it represented a big chunk of the available data for replication – there were a few cohorts not included, but they were generally difficult to access, or considerably smaller than this new GWAS (and so a failed replication might simply be because the replication cohort was too small to see anything. So how to replicate? Leave one out polygenic scoring showed promising results, but could the investigators go further? Obviously. Why else would I be bringing up this paper (other than the scientific brilliance, wit and sheer aura of raw physical attractiveness of the fifth-named author?). Sniekers and colleagues used another technique for replication, that of the proxy-replication.
Educational success is correlated with IQ. It is also relatively straightforward to measure. If you have a group of genotyped people, and permission to access their exam results (or to ask them how many years they were in education for), you can do a GWAS of educational attainment. This had in fact been very successful – the latest such GWAS had identified 74 regions. Looking up 306 variants that differed strongly with IQ in their data, the authors showed that 305 of these affected educational attainment in the same direction as in IQ, and that 9 of the 16 top regions they could test from the IQ differed in educational attainment more than would be expected by chance alone. When the exact same trait isn’t available, related traits can be informative.
3. Wait
GWAS are like buses. They carry a lot of people, and you tend to get on strange ones when you’re drunk. [Edit: What?! From context alone I can only assume you were actually going for “there’s always another one along in a minute”…]

A shiny GWAS
With time come new samples, new data and replication. Take the 18 IQ loci – in a cohort nearly four times the size, 15 of those loci remained significant. Or look at the schizophrenia region found in 2009 – that’s now arguably the most robust region in schizophrenia genetics, with a plausible functional role (that perhaps wasn’t the one initially expected) and is now coming up in other psychiatric disorders like depression (arguing that it might be associated with psychiatric disorders more generally). As such, a kind of replication reminiscent of the traditional method is still possible in GWAS, through building new cohorts into the older cohorts, and seeing what survives.
Because it’s now Christmas, as told by the traditional herald angels John and Lewis, I leave you with a present of blogs yet to come focussed on the spirit of GWAS past. Here, I’ve said a kind of replication can be achieved by the addition of new samples to older GWAS. Next time, I will show that it can be informative to look back at old GWAS to understand the evolution of newly-discovered hits.