 - Part 2")
Pre-clinical models, by Dr William Hill
This is the third in a special four-part series, looking at how air pollution can cause lung cancer in people who have never smoked.
Authors on the paper, Professor James DeGregori, Dr William Hill, and Dr Emilia Lim all contributed to the series.
Our previous blog post, by Dr Emilia Lim, discussed our research exploring the epidemiological relationship between EGFR-mutant lung cancer and levels of air pollutant PM2.5. Building on this, and hunting for these mutations in normal human lung tissue, we turned to the lab to understand how this might be happening.
We utilised pre-clinical models to explore this in more detail.
PM-mediated promotion of lung cancer
Firstly, we developed genetically engineered mouse models in which we could induce the oncogenic EGFRL858R mutation in mouse lungs. We exposed these to physiologically-relevant concentrations of fine particulate matter (PM): particles of air pollutants collected in cities. Using two parallel models, we found that exposure to PM increased numbers of early pre-invasive neoplasia and formation of adenocarcinoma.
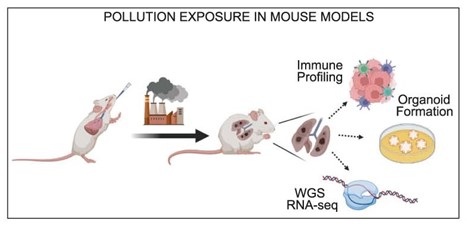
We next wanted to test whether there was a specific relationship between EGFR mutations and air pollutants. We investigated this in another mouse model, using expression of the oncogenic KRAS mutation. However, we found that PM also promoted KRAS-mutant lung cancer.
Immune-dependent nature of PM carcinogenesis
To test whether PM promotes lung cancer by inducing mutations, we carried out whole genome sequencing of mouse tumours, comparing mice with and without exposure to air pollutants. But we found no increase in mutations in the tumours from PM-exposed mice. This suggested that air pollution instead acts via another cellular mechanism.
To investigate the function and potential involvement of specific immune cells in mediating tumour promotion, we used ‘knockout mice’: mice lacking key parts of their immune system. When we repeated the same experiment in our immune-deficient (knockout) mice, we found that air pollutants no longer promoted early lung cancer. This demonstrated that immune cells play a key role in the promotion of early lung cancer.
We investigated the different immune cells changing upon exposure to PM. From this, we identified macrophages (immune cells important for detection and destruction of foreign organisms) as one of the major cell populations changing. It seems that air pollutants cause an influx of macrophages into the lung and a release of inflammatory cytokines.
But how does this promote lung cancer?
PM-mediated cell reprogramming
In healthy tissue, growth and proliferation of cells remains under strict control by a variety of immune and cellular mechanisms. This prevents cell outgrowth and cancer, and extends to keeping cell types specialised once differentiated. We therefore investigated whether cells are more undifferentiated (more progenitor-like) when exposed to air pollution. This was done using a ‘colony-forming assay’, in collaboration with our colleagues Prof Ilaria Malanchi and Felipe Rodrigues.
From profiling epithelia, we found evidence that exposure to PM was transcriptionally altering cells into a more progenitor-like state. We found that the combination of air pollutant exposure and EGFR-mutant cell types alone increased the number of organoids formed.
We next tested whether the key reprogramming signal for this was coming from macrophages. To do this, we harvested macrophages from mice exposed to air pollution and EGFR-mutant cells from animals never exposed to air pollution. We then cultured these together. From this, the number of organoids formed increased. This demonstrated that macrophage signalling is capable of reprogramming EGFR-mutant cells into a more progenitor-like state. Note: A ‘progenitor cell’ is only capable of differentiating into cells that belong to the same tissue or organ V’s a stem cell that can differentiate into multiple types of cells.
PM induces IL-1β production from macrophages
As a final step, we wanted to test if this macrophage signalling is important for lung cancer promotion. We firstly found that PM drives interleukin-1 beta (IL-1β) production in macrophages and epithelia. We also found that IL-1β drives the growth of EGFR-mutant organoids.
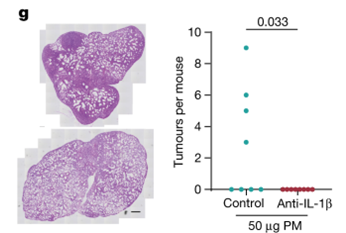
From this, we honed in on IL-1β. There has been related clinical trial evidence in which antibody targeting of IL-1β decreased the number of new lung cancer cases, in a dose-dependent fashion. We therefore wondered whether IL-1β might be a trigger for the promotion of lung cancer by air pollutants. To test this, we administered an anti-IL-1β antibody during exposure to PM.
We found that that this anti-IL-1β antibody suppressed tumour promotion in our models.
Key findings
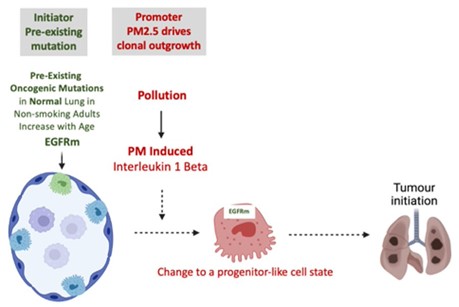
From these results, we have proposed that air pollutants stimulate an influx of macrophages into the lung and a release of IL-1β. This triggers a reprogramming of EGFR-mutant lung epithelial cells into a more progenitor-like cell state. This in turn fuels tumorigenesis.
This study has changed the way we think about how environmental exposures and carcinogens might drive cancer growth. It holds the potential for opening new avenues to identify people at higher risk of cancer, and even for developing therapies to prevent cancer.
These implications will be discussed in full in the final blog post of this series.
The views expressed are those of the author. Posting of the blog does not signify that the Cancer Prevention Group endorses those views or opinions.
Leave a Reply